From Chalkboards to Code: How Algorithms are Revolutionizing the Hunt for New Medicines
In the 1980s, scientists tackling the HIV epidemic spent nearly a decade developing azidothymidine (AZT)—the first antiretroviral drug. The process was slow, costly, and driven largely by trial-and-error experimentation. At that time, researchers had limited tools and had to test hundreds of molecules in the lab, one at a time, before identifying a promising candidate.
Today, that process has radically changed.
In 2020, during the early stages of the COVID-19 pandemic, researchers employed Computer-Assisted Drug Design (CADD) to model the structure of the SARS-CoV-2 main protease (Mpro), a key enzyme that facilitates the virus's replication. Within days, supercomputers screened millions of compounds in silico to identify molecules that could bind to and inhibit this enzyme. One such success story was Pfizer's Paxlovid, which includes nirmatrelvir, a molecule designed with heavy reliance on structure-based drug design.
What once took years now takes weeks, or even hours. This is the power of CADD!
By integrating quantum chemistry, bioinformatics, machine learning, and molecular dynamics, CADD platforms can simulate how a drug interacts with its target at the atomic level. It enables scientists to virtually test the "fit" between a drug and its receptor, optimize lead compounds, and even predict off-target effects or toxicity, long before a single animal study is done.

From pandemic responses to precision oncology, CADD is no longer a futuristic concept; now it is a core strategy in pharmaceutical innovation. But what are the techniques that drive CADD? What tools are used? And how are companies—from startups to big pharma—leveraging this technology to cut costs, reduce failures, and bring medicines to market faster?
This article explores the full landscape of Computer-Assisted Drug Design: from ligand-based and structure-based approaches to AI-driven platforms reshaping the future of drug discovery.
Connect with our scientific experts for your drug discovery, development and manufacturing needs
What is Computer-Assisted Drug Design (CADD)?
CADD is the use computational tools and methods that assist researchers in discovering and optimizing new drugs with greater precision and efficiency. By simulating molecular interactions and predicting biological behavior, CADD significantly reduces reliance on costly and lengthy laboratory experiments in early drug discovery phases.
CADD combines interdisciplinary insights from genomics, structural biology, and cheminformatics to significantly enhance the accuracy and efficiency of drug discovery processes. At its core, CADD systematically evaluates how molecular structures interact at an atomic and subatomic level, providing researchers with precise predictions and actionable insights to drive drug discovery forward.
i. Predicting drug–target binding interactions
CADD methodologies extensively utilize structural data obtained through high-resolution techniques such as X-ray crystallography, nuclear magnetic resonance (NMR), and cryogenic electron microscopy (cryo-EM). Using these structural insights, computational algorithms simulate how potential drug candidates fit into and interact with specific biological targets, such as proteins, enzymes, or receptors. This precise modeling enables researchers to anticipate binding strength, stability, and selectivity, significantly narrowing the scope of experimental validation required and greatly accelerating the identification of promising lead compounds.

ii. Anticipating off-target effects and toxicity
CADD approaches also consider the potential interactions of drug candidates with unintended biological targets. By computationally screening candidate molecules against large databases of known biological interactions, researchers can anticipate potential adverse reactions or toxicity issues early in the discovery pipeline. These in silico toxicity predictions help avoid costly late-stage drug failures and enable refinement of lead compounds toward safer, more selective profiles before committing substantial resources to experimental testing.
iii. Modeling pharmacokinetics and pharmacodynamics (PK/PD)
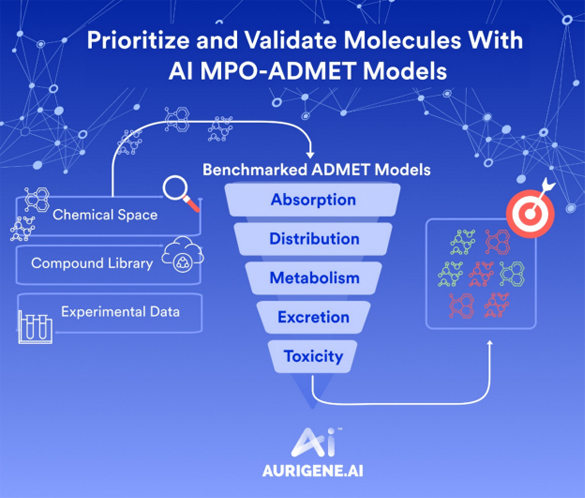
Beyond interaction studies, CADD tools can forecast how a candidate molecule will behave in the human body. Using extensive computational modeling, algorithms can predict essential pharmacokinetic properties, such as absorption rates, distribution patterns, metabolic stability, excretion pathways, as well as pharmacodynamics, or how drugs exert their effects within the body. By simulating these dynamic biological processes, researchers gain valuable early insight into dosing regimens, therapeutic windows, and potential efficacy, significantly optimizing drug development strategies.
Together, these core principles represent a fundamental shift in drug discovery philosophy. Rather than relying primarily on trial-and-error methodologies or intuition, CADD promotes a structured, rational approach based on systematic computational analysis. This transition dramatically reduces development timelines, cuts costs, increases precision, and improves the probability of successful clinical translation, positioning computational tools as essential components of modern pharmaceutical innovation.
CADD broadly employs two main strategies: structure-based and ligand-based design, each addressing unique scenarios in drug discovery.
Structure-Based Drug Design (SBDD) utilizes detailed three-dimensional structural data of the biological target, typically derived from advanced techniques such as X-ray crystallography or cryo-EM. Based on this knowledge, researchers perform molecular docking simulations to predict how potential drug molecules align and interact within the binding pockets of proteins. These docking simulations provide insights into the optimal positioning, orientation, and shape complementarity between drugs and their targets. Further, molecular dynamics simulations are employed to analyze the dynamic behavior and stability of the drug-target complexes, assessing how well a candidate might remain bound under physiological conditions. To quantitatively measure these interactions, computational free energy calculations estimate binding affinities and strengths, thereby identifying compounds with the highest therapeutic potential.
By contrast, Ligand-Based Drug Design (LBDD) is primarily employed when the precise structural details of the biological target are not known. In such cases, computational models rely on the known chemical structures and biological activities of previously characterized active molecules. Pharmacophore modeling, a key ligand-based method, involves identifying essential structural and chemical features common to active compounds, thereby guiding the design of new molecules with similar or improved biological activities. Additionally, Quantitative Structure-Activity Relationship (QSAR) analyses statistically correlate molecular structural features with observed biological effects, enabling the prediction of biological activity for novel compounds. Another significant ligand-based technique, similarity searching, rapidly screens extensive chemical databases to identify compounds structurally or chemically analogous to established active molecules, accelerating the discovery and optimization of new drug candidates.
Together, these complementary approaches empower researchers to strategically navigate complex chemical spaces, efficiently pinpointing molecules with enhanced therapeutic properties and significantly streamlining drug development timelines.
CADD has fundamentally reshaped pharmaceutical discovery, driving breakthroughs that have led to significant advances in medical treatments. Several landmark drugs stand as testaments to the profound impact and practical success of computational methods.
Imatinib (Gleevec):
Imatinib, commercially known as Gleevec, exemplifies the transformative potential of rational drug design, a cornerstone of CADD methodologies. Its development was specifically guided by an in-depth computational understanding of the BCR-ABL kinase, an abnormal fusion protein responsible for chronic myeloid leukemia (CML). Researchers employed precise computational modeling and structure-based drug design techniques to create molecules tailored to fit snugly into the ATP-binding site of this oncogenic protein. By selectively targeting BCR-ABL, imatinib dramatically improved patient outcomes, elevating five-year survival rates significantly and transforming a once-fatal condition into a manageable chronic illness. The rational design approach facilitated by computational methods dramatically reduced the time and resources required compared to traditional drug development processes.
HIV protease inhibitors:
The battle against HIV/AIDS also benefited greatly from advancements in CADD. In the early stages of the epidemic, therapeutic options were limited, and discovery timelines were prolonged due to reliance on experimental screening methods. Computational tools changed this trajectory. By leveraging detailed structural insights obtained through techniques such as X-ray crystallography, researchers were able to precisely model HIV protease—a critical viral enzyme essential for HIV replication. Structure-based drug design methods, including molecular docking and dynamics simulations, enabled rapid screening and optimization of potential inhibitors. Consequently, potent HIV protease inhibitors such as ritonavir, saquinavir, and indinavir were developed much faster, drastically altering HIV management. These computationally derived treatments transformed HIV/AIDS from a fatal diagnosis into a chronic, manageable condition, highlighting the profound societal impact of computational approaches in pharmaceutical research.
Selective COX-2 inhibitors:
Another significant success attributed to CADD is the development of selective cyclooxygenase-2 (COX-2) inhibitors, exemplified by celecoxib. Historically, anti-inflammatory drugs like traditional non-steroidal anti-inflammatory drugs (NSAIDs) inhibited both COX-1 and COX-2 enzymes, often causing severe gastrointestinal complications. Using computational modeling and structure-based analyses, researchers identified subtle differences between COX-1 and COX-2 active sites. Advanced computational screening techniques helped pinpoint molecular structures selectively interacting with COX-2 while minimally affecting COX-1. This selective inhibition dramatically reduced gastrointestinal side effects and improved the safety profile of anti-inflammatory therapies. Celecoxib, a widely used drug developed through this method, exemplifies how CADD can systematically enhance drug efficacy while simultaneously mitigating adverse side effects, offering safer therapeutic options to millions of patients globally.
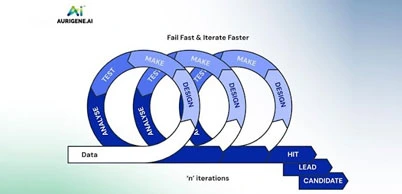
Accelerated discovery through high-speed virtual screening
Traditional drug discovery methods, reliant heavily on extensive laboratory experimentation, frequently involved manually testing vast arrays of chemical compounds—a slow, resource-intensive process that could take years. CADD significantly accelerates this stage by enabling rapid computational screening of millions—or even billions—of molecular structures within hours or days. Advanced computational algorithms, utilizing molecular docking, pharmacophore mapping, and machine learning-driven predictive modeling, efficiently identify molecules with high potential for biological activity. By quickly eliminating unsuitable compounds from consideration, computational screening allows researchers to focus laboratory resources exclusively on the most promising drug candidates, significantly shortening the early stages of the drug discovery timeline.
Cost-efficiency through strategic reduction in experimental work
A major financial bottleneck in drug development arises from extensive experimental trials, synthesis, and biological testing of numerous candidate molecules. CADD substantially reduces these costs by systematically narrowing down chemical libraries to fewer, more strategically selected molecules before laboratory validation. Computational methods effectively predict toxicity profiles, binding affinities, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties early in the discovery process, preventing expenditure on compounds likely to fail later-stage tests. As a result, companies save significant resources, minimize financial risks, and optimize their research and development budgets, enabling smaller biotech firms and academic groups to participate actively in innovative drug discovery.
Unparalleled precision at the molecular level
CADD enables researchers to explore molecular interactions with unprecedented detail and accuracy. Utilizing advanced quantum chemical calculations, molecular dynamics simulations, and accurate free-energy perturbation techniques, computational models precisely simulate drug-target interactions at atomic resolution. This precision empowers researchers to understand exactly how slight modifications in chemical structure influence biological activity, binding affinity, selectivity, and stability. Such atomic-level insight is invaluable for designing molecules with highly specific and potent biological actions, significantly reducing unintended interactions or adverse effects. Consequently, drugs developed through CADD typically exhibit enhanced efficacy, improved safety profiles, and optimized pharmacokinetic characteristics, underscoring the transformative precision this approach offers.
Increased success rates through enhanced lead optimization
Historically, many promising drug candidates fail late in development due to unforeseen toxicities, suboptimal pharmacological properties, or inadequate efficacy profiles. CADD addresses these challenges by thoroughly vetting candidate molecules through comprehensive computational analysis prior to advanced experimental stages. Computational predictions inform lead optimization, enabling rapid iterative refinement cycles where molecular modifications can be immediately assessed and validated virtually. This rigorous pre-experimental vetting markedly decreases the likelihood of late-stage failures, thereby enhancing overall drug development success rates. As a direct consequence, drug candidates entering clinical trials often exhibit stronger efficacy and safety profiles, improving regulatory approval rates and expediting their availability to patients.

Recent advancements have profoundly broadened the potential of CADD, transforming drug discovery from a traditional trial-and-error methodology into a sophisticated computational science.
AI and deep learning: unlocking hidden patterns and predictive power
Artificial intelligence (AI), particularly deep learning, has dramatically enhanced the capabilities of computational drug discovery. Deep neural networks trained on extensive datasets containing millions of protein-ligand interactions can identify subtle molecular patterns and intricate biological interactions often invisible to human observation or conventional computational methods. AI-driven algorithms now predict novel molecular structures that exhibit optimal pharmacological profiles, generating entirely new classes of therapeutic compounds. By harnessing machine learning, researchers rapidly predict efficacy, selectivity, and potential side effects, facilitating more targeted, informed drug design. These techniques have already accelerated drug discovery for challenging targets previously considered inaccessible due to complexity or data scarcity.
Cloud-based Platforms: Facilitating Global Collaboration and Rapid Data Sharing
Innovations in cloud computing have revolutionized collaborative drug discovery, enabling instantaneous sharing of complex computational analyses, vast datasets, and predictive modeling across international research teams. Cloud-based drug design platforms enable real-time collaboration, democratizing access to sophisticated computational resources previously available only to major institutions with substantial computational infrastructure. This global collaboration approach shortens research timelines by streamlining the sharing and validation of computational results, fostering rapid iteration of molecular designs, and improving reproducibility of scientific findings. Such platforms significantly accelerate preclinical discovery and facilitate swift, coordinated responses to urgent public health challenges.
Antimicrobial Drug Discovery: Precision Targeting via Computational Modeling
The CHARM (Center for Host-Microbe Systems and Therapeutics) initiative at UC San Diego exemplifies a practical and highly impactful application of CADD by addressing the critical public health threat of antibiotic resistance. Using advanced computational modeling, researchers have precisely designed narrow-spectrum antimicrobial compounds that specifically target harmful pathogens without affecting beneficial microbiome communities. This approach minimizes the collateral damage traditionally associated with broad-spectrum antibiotics, potentially reducing antibiotic resistance and preserving microbiome health. Such precision-targeted antibiotics represent a critical innovation in managing infections effectively while mitigating long-term public health consequences.
COVID-19 Therapeutics: Rapid Response Enabled by Computational Predictions
The rapid development of Pfizer's antiviral Paxlovid, which includes the computationally identified active component nirmatrelvir, illustrates the immense value of CADD in addressing global public health crises. In the early stages of the COVID-19 pandemic, computational tools were swiftly deployed to identify and validate candidate molecules capable of binding effectively to SARS-CoV-2 viral proteins, particularly the main protease (Mpro). Computational simulations accurately predicted optimal binding interactions, significantly accelerating the transition from theoretical modeling to laboratory synthesis and clinical testing. This accelerated pathway enabled unprecedented speed in bringing critical antiviral therapies to global populations, underscoring CADD's immense public health value.
Challenges and Limitations of CADD
Despite remarkable successes, computational drug design methodologies face intrinsic constraints that must be acknowledged and carefully managed.
Data Quality: The Essential Foundation of Reliable Predictions
CADD's predictive accuracy hinges fundamentally upon the quality of the data used for computational modeling. Erroneous, incomplete, or inconsistent structural and biochemical data directly compromise model accuracy, leading to unreliable or misleading predictions—often encapsulated in the cautionary principle of "garbage in, garbage out." Consequently, rigorous data validation, meticulous database curation, and standardized data collection procedures are essential for reliable computational modeling. Researchers must continually refine computational methodologies and data collection practices to enhance predictive reliability.

Biological complexity: capturing reality through computational models
While sophisticated, current computational models often struggle to fully encapsulate the dynamic complexity inherent to biological systems. Proteins in vivo exhibit flexibility, conformational variability, and intricate interactions influenced by solvents, ions, and cellular environments—complexities that remain challenging to accurately represent computationally. Moreover, metabolic transformations, drug transport, and interactions with multiple molecular pathways complicate the accurate computational prediction of a molecule's biological fate. Overcoming these limitations requires advanced methods integrating quantum mechanics, multi-scale modeling, and high-performance simulations, complemented by experimental validation.
Integration with experimental validation: bridging computational predictions and reality
CADD findings must always be integrated with and validated through rigorous experimental testing. Although computational methods greatly streamline initial discovery phases, they cannot fully replace laboratory validation. Computational predictions serve best as a complementary tool, providing hypotheses for subsequent experimental verification. Seamless integration between computational predictions and experimental validation methods, such as biochemical assays, cell-based experiments, and in vivo models, is crucial. The synergy between computational methods and laboratory experimentation remains fundamental to drug discovery.
Comprehensive CADD Services Offered by CDMOs
- Structure-based drug design (SBDD): Employing high-resolution structural data, such as X-ray crystallography or cryo-EM, to model the three-dimensional structure of target proteins. This approach facilitates the design of compounds that fit precisely into the active sites of these targets, enhancing binding affinity and specificity.
- Ligand-based drug design (LBDD): Utilizing information from known active compounds to predict the activity of new molecules. Techniques like Quantitative Structure-Activity Relationship (QSAR) modeling and pharmacophore mapping help identify key molecular features responsible for biological activity, guiding the design of novel therapeutics.
- Virtual screening: Rapidly evaluating large libraries of compounds using computational methods to identify potential drug candidates. This process prioritizes molecules with favorable properties for further experimental validation, significantly reducing time and resources.
- De novo design & scaffold hopping: Creating entirely new molecular structures or modifying existing ones to improve efficacy, selectivity, or pharmacokinetic properties. This strategy explores diverse chemical spaces to identify innovative therapeutic candidates.
- Molecular docking and binding mode prediction: Simulating the interaction between small molecules and their biological targets to predict binding orientations and affinities. This insight aids in optimizing compound structures for enhanced activity.
- Pharmacophore modelling: Identifying the essential features of molecules responsible for their biological activity. This model serves as a template for designing new compounds with similar or improved therapeutic effects.
- Homology modelling: Constructing three-dimensional models of target proteins based on the known structures of related proteins. This is particularly useful when experimental structures are unavailable, enabling structure-based design approaches.
- ADMET predictions: Assessing the Absorption, Distribution, Metabolism, Excretion, and Toxicity profiles of compounds using computational tools. Early prediction of these properties helps in selecting candidates with favorable drug-like characteristics.
- Druggability assessment: Evaluating the likelihood that a biological target can be modulated by a drug-like molecule. This assessment informs target selection and prioritization in drug discovery programs.
- Quantum mechanics / molecular mechanics (QM/MM) analysis: Applying hybrid computational techniques to study molecular interactions at both quantum and classical levels. This provides detailed insights into reaction mechanisms and binding energetics.
- Molecular dynamics simulation: Modeling the physical movements of atoms and molecules over time to understand the stability and conformational changes of drug-target complexes under physiological conditions.
- In silico lead optimization: Iteratively refining lead compounds using computational methods to enhance potency, selectivity, and pharmacokinetic properties before synthesis and experimental testing.
- Integration with medicinal chemistry and biology: Collaborating closely with medicinal chemists and biologists to ensure that computational predictions align with synthetic feasibility and biological relevance, streamlining the drug development process.
What Aurigene offers
Computational Infrastructure:
- High-performance NVIDIA GPU clusters and workstations
- Access to Google Cloud for scalable computing needs
Modeling and Simulation Platforms:
- Nanome interactive virtual reality platform
- Molsoft and Schrödinger Suite (commercial software)
- Open-source tools: KNIME, Cytoscape, DataWarrior
Databases:
- Access to public domain biological and chemical databases
- In-house Aurimine database focused on target-relevant compounds
- Multi-billion compound commercial databases for virtual screening
CADD Core Services:
- Virtual Screening: Hit identification using ligand- and structure-based methods, GIGA-scale virtual screening
- Structure-Based Drug Design: Docking, molecular dynamics (microsecond–millisecond scale), MMGBSA, FEP
- Ligand-Based Drug Design: Pharmacophore modeling, QSAR, scaffold hopping, ligand hybridization, fragment linking
- De Novo Design & Lead Optimization: AI/ML-driven predictive models for potency, selectivity, and drug-likeness
Informatics Services:
- Target analysis and next-generation sequencing (NGS) analysis
- Systems biology, structural bioinformatics, and pathway/network modeling
- Sequence database manipulation and protein structure prediction
- Ontology mapping, graph/network analysis, clustering, and enrichment analysis
ADMET & PK/PD Prediction:
- In silico pharmacokinetic and pharmacodynamic profiling
- Machine learning-based ADMETox models and profile QSAR methods
Therapeutic Modalities Supported:
- Small molecules and peptides
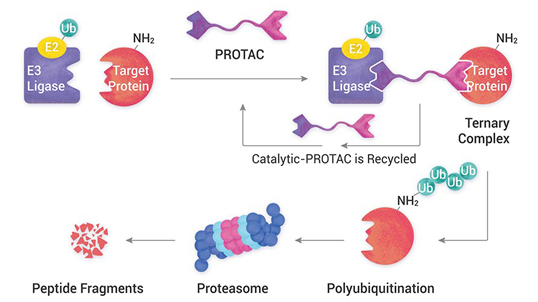
- Targeted protein degradation (PROTACs, molecular glues)
- Antibody and ADC (antibody-drug conjugate) design
- Oligonucleotides (e.g., siRNA, aptamers)
- mRNA vaccine optimization
Scientific Capabilities by Modality:
- Peptides: Generative design, protein–protein interaction modeling, property optimization
- Biologics: Protein humanization, de novo design, off-target prediction
- Oligonucleotides: Aptamer and siRNA design, structure-based small molecule design
Collaborative Model:
- Integrated discovery approach from target ID to candidate nomination
- Coordination between computational chemists, experimental biologists, and medicinal chemists
- Emphasis on translational biology and project-specific ideation support
Team Expertise:
- Multidisciplinary team of highly trained scientists
- Proven capability in solving complex drug discovery problems
- Demonstrated success in niche therapeutic programs
Future of CADD: Embracing Artificial Intelligence and Advanced Computational Techniques
Looking forward, innovations in AI and advanced computational methodologies promise to further revolutionize CADD, enhancing both the scope and accuracy of drug discovery.
Predictive and generative AI: redefining molecular discovery
Advanced AI methodologies, particularly predictive and generative deep learning models, are set to fundamentally redefine drug discovery paradigms. Predictive AI algorithms trained on extensive experimental data will increasingly accurately forecast drug-target interactions, binding affinities, and biological activities. Concurrently, generative AI, employing techniques such as generative adversarial networks (GANs) and reinforcement learning, can automatically propose novel molecular structures optimized for therapeutic effectiveness and reduced side effects. Such technologies expand chemical space exploration far beyond conventional boundaries, enabling the design of innovative therapeutic agents with unprecedented speed and accuracy.
Personalized therapeutics: computational tools tailored to individual genetics
The integration of AI-driven computational drug design into personalized medicine represents a particularly exciting frontier. Advanced computational models, combined with individualized genomic, proteomic, and metabolomic data, will increasingly enable the tailored design of therapeutic strategies customized to individual patients' genetic and molecular profiles. By predicting individualized drug responses, side effects, and optimal dosing regimens computationally, AI-supported personalized medicine approaches can maximize therapeutic efficacy, minimize adverse reactions, and improve overall patient outcomes significantly. This approach represents a profound shift from traditional one-size-fits-all treatment paradigms toward more precise, individualized care.
In conclusion, continuous advancements in artificial intelligence, enhanced modeling of biological complexity, improved data quality, and strengthened integration with experimental methodologies position CADD at the forefront of drug discovery. By embracing these technological developments and addressing intrinsic limitations, computational drug design methodologies will continue shaping pharmaceutical research and therapeutic innovation for decades to come.