
In the early 2000s, developing Sovaldi, a hepatitis C treatment, took over a decade and nearly $2 billion. Similarly, Zolgensma, a gene therapy for spinal muscular atrophy, required 15 years due to its complexity. However, the advent of artificial intelligence (AI) has revolutionized drug discovery. For example, in 2022, Pfizer's PAXLOVID, an oral COVID-19 treatment, was developed in just four months, reducing computational time by 80-90% and costs to $500 million. Exscientia's AI-driven OCD drug reached clinical trials in 12 months, costing about $50 million. BenevolentAI identified Baricitinib for COVID-19 within months, repurposing an existing drug at lower costs. Insilico Medicine discovered a new drug candidate for idiopathic pulmonary fibrosis in 18 months, with development costs of around $70 million, and it has successfully completed Phase IIa clinical studies. Additionally, Insilico Medicine claimed to have achieved 22 preclinical candidates in 3 years, with an average timeline of 13 months and around 70 molecules synthesized per integrated drug discovery (IDD) program. These examples highlight the transformative power of artificial intelligence (AI) in accelerating drug discovery, reducing time and cost, and making lifesaving treatments more accessible and affordable. The AI revolution is not just about automation anymore; it is about autonomous intelligence systems allowing AI to make decisions, take actions, and continuously learn with minimal human input on complex discovery tasks. These autonomous AI systems will help enhance efficiency, improve customer experiences, and support revenue growth in the pharmaceutical business.
Limitations of Traditional Drug Discovery
Traditional drug discovery is labor-intensive, costly, and heavily reliant on trial-and-error. The process can take 2.5 to 4 years to reach IND filing, while developing a new drug can take 12 to 15 years and nearly $2 billion. Increasing R&D expenses and high failure rates in late-stage clinical trials pose significant challenges. Many potential compounds fail due to poor pharmacokinetics and pharmacodynamics, limited understanding of disease mechanisms, and inadequate preclinical models. Balanced in vitro potency, ADME (absorption, distribution, metabolism, and excretion), toxicity, and physicochemical properties are crucial for successful drug development. New approaches are needed to determine the efficacy and safety of molecules and generate high-quality drug-like molecules with desired on-target potency, off-target selectivity, ability to reach the target site, intended pharmacological effects, and suitable pharmaceutical properties for synthesis, formulation, and dosage regimen.
Emergence of AI in Discovery
Over the past three decades, computational chemistry has made significant progress in drug discovery through structure-based and ligand-based virtual screening approaches. However, conventional computational techniques like library enumeration or the utilization of pre-constructed virtual libraries investigate only restricted areas of chemical space. Lead optimization is often done sequentially, starting with potency, then selectivity, and subsequently pharmacokinetic attributes. This approach has resulted in a less-than-ideal process that failed to substantially accelerate discovery timelines. Additionally, the unpredictable nature of synthesis timelines introduces bottlenecks that impede the overall efficiency of the Design-Make-Test-Analysis (DMTA) cycle, thereby delaying progress in drug discovery.
To overcome these challenges, AI has emerged as a transformative tool, opening the door to the next frontier in drug discovery by enhancing data processing, pattern recognition, predictive analytics, and generative tasks. The integration of AI in drug discovery is revolutionizing the pharmaceutical industry by streamlining protein and molecular design, exploring ultra-large chemical space for novel therapies, automating synthesis, and enhancing the overall quality and speed of discovery. Generative AI emerged as a game-changer in 2024, enabling scientists to generate novel molecules beyond existing compounds and datasets and to discover potential novel therapeutic targets. Many generative AI models incorporate user-specified scaffolds, predicted and computed properties, drug-likeness filters (QED), toxicity alerts, synthetic accessibility filters, evaluation metrics (novelty, uniqueness, validity, similarity, diversity), AI-based molecular docking, FEP and MDS calculations for prioritizing the right compounds, which can be made and tested quickly.
Recent AI Breakthroughs in Pharma:
Recent advancements in AI over the past couple of years have significantly impacted drug discovery and personalized medicine. In 2025, agentic AI, multimodal AI, self-supervised learning, and federated learning have accelerated drug discovery processes, enhanced data privacy, and addressed data scarcity. Virtual cell simulations, digital twins, and generative AI have enabled personalized medicine and novel protein design. In 2024, notable breakthroughs included Nobel Prizes awarded for computational protein design and machine learning with neural networks, AI-powered digital twins for personalized disease models, and zeroshot generative AI in antibody design. MIT's DiffDock, NVIDIA's BioNeMo Cloud, and FDA clearance of AI-discovered TACC3 PPI inhibitor for women's cancer were also significant. In 2023, advancements included Deep Genomics' predictions for oligonucleotides, Google DeepMind's protein 3D structure predictions, Insilico Medicine's GAN for DDR1 inhibitions, RoseTTAFold's rapid protein structure calculations, and the application of large language models in drug discovery. These developments highlight the transformative impact of AI in making drug discovery faster, more efficient, and precise.
Impact of AI on drug discovery DMTA cycle:
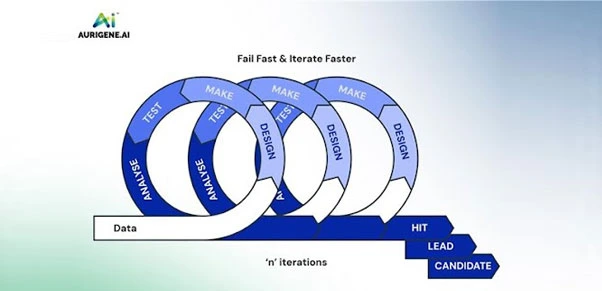 Figure: Identification of Hit, Lead and Candidate through iterative DMTA cycle
Figure: Identification of Hit, Lead and Candidate through iterative DMTA cycleDMTA is an iterative cycle that involves designing, making, testing, and analysing compounds. AI in DMTA cycle facilitates hit-to-lead identification and lead optimization by generating new molecules, exploring new chemical space, and predicting their potency, efficacy, toxicity, and pharmacokinetics through multi-parametric approaches. It automates molecule synthesis and integrates assay results and predictions into subsequent design iterations. Additionally, AI supports retrosynthetic planning, including the prediction of synthetic routes, cost, and availability of building blocks, ordering of necessary reagents and materials, prediction of reaction conditions, reaction impurities, yield of the product, catalyst design and reaction optimization, and de novo synthetic reaction design. The de novo molecular design and its optimization in parallel enhance decision-making for medicinal chemists and biologists, enabling them to select the right compounds throughout the DMTA cycle and between cycles, thereby reducing the time and cost of the discovery process.
Trends in AI-Driven CROs & CDMOs:
The AI-enabled drug discovery landscape within the contract research organization (CRO) and contract development and manufacturing organization (CDMO) sectors is evolving rapidly and playing a critical role in driving innovation and efficiency across the industry. The global pharmaceutical CRO market was valued at approximately $41.22 billion in 2024 and is projected to grow at a CAGR of 7.39% from 2025 to 2030 (https://www.grandviewresearch.com/industry-analysis/pharmaceutical-cro-market-report). The AI-driven services CRO and CDMO market is expected to reach $338.13 billion by 2030 (https://www.credenceresearch.com/report/pharmaceutical-cro-and-cdmo-market). Reports demonstrate that the CRO market is seeing a rise in strategic collaborations and partnerships between AI-driven drug discovery firms and pharmaceutical companies. Currently, over 270 AI biotech companies, 1500 investors, and 120 pharmaceutical companies are utilizing AI in their research. As of now, there are no drugs that have been approved by the FDA solely based on AI. However, AI has significantly contributed in identifying more than 250 preclinical and 25 clinical candidates. Global reports indicate that AI is making a significant impact by reducing time by 40%, costs by 10%, and increasing accuracy by 30%. Most AI-native companies are focusing on metabolic and cardiovascular diseases, oncology, respiratory, and anti-infective diseases.
What is Aurigene.AI?
Aurigene.AI is a web-based cloud platform designed to accelerate the drug discovery process by reducing the time required for hit identification and hit-to-lead optimization through generating new molecules and predicting drug-like ADMET properties. It is a secure and scalable platform hosted in Google cloud platform that offers research as a service (RaaS) for customers in biotech and pharma companies. Aurigene.AI processes vast datasets quickly, identifies patterns that might be missed by traditional methods, and predicts outcomes like drug efficacy and toxicity for reducing the risk of late-stage failures and lowers overall costs. It is user-friendly no-code platform to the medicinal chemists for design and prioritization of new molecules, optimization of structure-activity relationship towards lead optimization.
The platform supports early preclinical discovery by enhancing the speed, accuracy, and cost-efficiency of the DMTA discovery cycle and between the cycles time. It analyses vast datasets quickly, train the models for identification of patterns that might be missed by traditional methods, and predicts outcomes like drug efficacy and toxicity for reducing the risk of late-stage failures and lowers overall costs. The platform is enabled with advanced deep generative models, multi-parameter optimization with explainable ML models, drug-likeness and structural alert filters, AI-based molecular docking along with traditional physics-based CADD models including free-energy perturbation (FEP) and molecular dynamics simulations (MDS). The ML models were benchmarked and lead within top 3 of the global models with respect to statistical parameters of MAE & AUROC, AUPRC. It offers a large library of 180 million real compounds, 10 billion synthesizable virtual compounds, 1.6 million bioassays and 0.18 million target libraries with structure search capabilities. The platform also integrated with structure normalization rules (SNRs) to enhance data quality and standards, enabling the generation of chemically valid molecules that can be utilized for both de novo molecular design and predictive modeling. An internal proof-of-concept (POC) case study demonstrated that Aurigene.Al supported 30% to 40% reduction of time in DMTA cycle and between the cycles of discovery process from hit identification to lead optimization.
How Aurigene.AI Streamlines the discovery:
Aurigene.AI mitigates AI risks by fostering collaboration between AI experts and scientists to bridge knowledge gaps and integrate multidisciplinary knowledge in biology, chemistry, and pharmacology. It ensures data quality and quantity and continuously expands in both public and commercial datasets to address data availability issues. The platform incorporates structure normalization rules to maintain data standards and leverages advanced AI algorithms to manage the complexity of chemical & biological systems. Aurigene.AI promotes interdisciplinary collaboration, develops interpretable AI models for better decision-making, and uses scalable cloud infrastructure to reduce costs and resource requirements. It integrates AI with traditional drug discovery methods for a holistic approach, employs an iterative DMTA cycle to enhance validation and reproducibility, and implements stringent data privacy and security measures to comply with regulatory guidelines.
Aurigene.AI’s future roadmap:
Aurigene.AI’s future roadmap is focused on creating an autonomous platform driven by agentic AI systems. This platform will seamlessly integrate design, optimization, and synthetic planning workflows, providing continuous feedback throughout the DMTA cycle to enhance decision-making for medicinal and computational chemists and biologists. It will support multidisciplinary integration across chemistry, biology, pharmacology, and clinical trials through a multi-agent system, utilizing advanced interpretable and explainable AI/ML models for both small and large molecules. Aurigene.AI’s multi-agentic AI systems are expected to manage integrated complex tasks such as target identification, drug repurposing, discovering new therapeutic indications, protein structure prediction, druggability assessment, biological pathway and protein-network analysis, compound screening, retrosynthesis prediction, toxicity prediction, and clinical biomarker prediction for the identification of drug-like lead molecules. The platform's capabilities can be further extended to the de novo design of proteins, polymers, oligonucleotides, peptides, PROTACs, molecular glues, and antibody-drug conjugates, ensuring a comprehensive end-to-end pipeline for drug discovery and development, from initial target identification to candidate nomination.
About the Author:

Dr. Janardhan is a seasoned data scientist with over 18+ years of experience in the pharmaceutical industry, specializing in AI, Data Science, and Informatics. He has worked for several pharmaceutical and IT companies, contributing to the strategic development of AI-enabled drug discovery platforms and managing pharma R&D projects related to data science. His expertise includes developing AI/ML capabilities specific to Big Data analytics, Predictive and Generative AI, Natural Language Processing, Deep learning, Large language models, Knowledge Graphs, Agentic AI systems, MLOps specific to delivering CI/CD pipelines, dockerized applications, and model deployments in a cloud environment.
Latest Posts
Transforming Drug Discovery with Aurigene.AI

ADMET Predictive Models in Aurigene.AI
PROTACs: Research for a life without cancer
Good practices in non-clinical toxicology assessment to accelerate IND and NDA Submissions
Continuous Flow Chemistry: A Game-Changer for Pharmaceutical Production

Why meet us at CPHI Milan?

Advancement in personalized medicine and how the CRDMO industry is part of the solution

Immuno Oncology - Therapies and Challenges

Building successful long-term partnerships with CDMOs from early drug discovery through commercialization
You are about to leave Aurigene Pharmaceutical Services and affiliates website. Aurigene Pharmaceutical Services assumes no responsibility for the information presented on the external website or any further links from such sites. These links are presented to you only as a convenience, and the inclusion of any link does not imply endorsement by Aurigene Pharmaceutical Services.
If you wish to continue to this external website, click Proceed.


Leaving already?
Don't forget to join us at
CPHI Worldwide 2023.
October 24th-26th, 2023 | Barcelona, Spain
Get ready to accelerate your drug’s journey to the market