DSP-1181 and the Rise of AI in Drug Discovery
In late 2019, a quiet revolution began in a laboratory in Oxford. Scientists at Exscientia, in collaboration with Sumitomo Dainippon Pharma, accomplished something unprecedented: they designed a novel drug candidate, DSP-1181, using artificial intelligence, and did it in just 46 days! The compound, a serotonin 5-HT1A receptor agonist for treating obsessive-compulsive disorder (OCD), took only a fraction of the time traditional drug discovery demands to progress from initial molecular design to preclinical testing.

The most important point to note is this was not just a faster process; rather, it was a fundamental reimagining of how drugs could be discovered. By leveraging deep learning, generative design, and predictive modeling, the AI system sifted through billions of potential molecular combinations, prioritized drug-like candidates, and optimized properties like potency, selectivity, and synthetic accessibility.
DSP-1181 was the first AI-designed molecule to enter clinical trials, and it signalled a new era. As the world grappled with the outbreak of COVID-19 shortly afterward, AI-assisted discovery moved from proof-of-concept to urgent necessity. In London, BenevolentAI scanned vast biomedical datasets using AI-driven knowledge graphs and identified baricitinib, a rheumatoid arthritis drug, as a promising candidate to counteract COVID-induced inflammation. Within weeks, baricitinib entered clinical trials and later received emergency use authorization. This is an example of an AI-guided repurposing success that saved time, money, and lives.
In Toronto, Cyclica deployed its Ligand Express platform to match existing molecules with viral targets, discovering potential antivirals against SARS-CoV-2 in record time by modeling how drugs interact with multiple targets simultaneously—a critical step in treating complex diseases.
Meanwhile, in China, Insilico Medicine applied generative adversarial networks (GANs) to create thousands of new molecular structures in under 72 hours. These were tailored to bind SARS-CoV-2's 3CL protease, a key viral replication enzyme. AI ranked them by drug-likeness and binding potential, reducing what once took months into days.
However, these were not isolated victories. They were the early signals of a tectonic shift. AI had begun transforming every phase of drug discovery—from molecular generation and virtual screening to preclinical optimization and repurposing.
Connect with our scientific experts for AI-powered drug discovery solutions
Overview of AIDD
Artificial Intelligence-Assisted Drug Discovery (AIDD) refers to the application of AI technologies such as machine learning, deep learning, natural language processing (NLP), generative models, and graph-based systems to enhance, accelerate, and optimize every stage of the drug discovery and development pipeline. These tools process enormous amounts of both structured and unstructured data—from chemical structures and omics datasets to clinical trial results and real-world patient information—and use that to build predictive models. These models help scientists to make informed decisions at every stage, from identifying drug targets and generating leads to optimizing compounds, validating them preclinically, and designing clinical trials.
AI models not only automate routine analyses but also surface hidden relationships between molecules, pathways, and disease phenotypes that are beyond human detection, and thus, offer a data-driven, adaptive framework that complements human expertise and significantly improves the efficiency and success rate of drug development efforts.
Why AIDD Matters
Drug development is an infamously slow, expensive, and failure-prone process, which often exceeds $2.6 billion in costs and takes more than a decade to bring a single drug to market. Most drug candidates fail during late-stage clinical trials, typically owing to issues related to efficacy, safety, toxicity, or suboptimal pharmacokinetics. AIDD is redefining this paradigm by introducing data-driven precision, automation, and adaptivity into the pipeline. AI enables the early identification and elimination of poor-performing compounds through predictive modeling, thus reducing the financial and time burden of downstream failures. It accelerates virtual screening, lead optimization, and synthetic route planning, significantly shortening the time from hit to lead.

In clinical development, AIDD enhances trial design through patient stratification, biomarker-driven enrolment, and predictive outcome modeling, substantially improving success rates. Furthermore, AI excels at revealing novel therapeutic uses for existing drugs, especially critical during public health emergencies such as pandemics. In addition to efficiency, AIDD introduces a more agile, scalable, and economically sustainable discovery framework that supports both high-throughput pipelines in big pharma and targeted therapies in rare disease research.
Core Technologies Powering AIDD
Machine Learning (ML)
Machine learning is becoming an essential tool in drug discovery, especially in predicting important molecular properties like binding affinity, toxicity, solubility, and permeability. Tree-based models and ensemble techniques, like random forests and gradient boosting, are often at the core of these predictions. Beyond that, machine learning also helps scientists make sense of high-throughput screening data by narrowing down the most important features and reducing the complexity of the data. A newer and promising direction is the use of self-supervised learning, which is helping models perform better even when there's not a lot of labeled data. This approach is proving especially useful for making molecular property predictions more reliable and broadly applicable.
Deep Learning (DL)
Deep learning models, such as convolutional neural networks (CNNs) for image-based assays and recurrent neural networks (RNNs) for sequential data, excel in extracting features directly from raw inputs. Graph-based deep learning, including message-passing neural networks (MPNNs), captures topological information in molecules, enabling accurate predictions of reaction outcomes and synthetic accessibility. DL is also crucial in analyzing complex imaging data, such as histopathological slides or phenotypic screens, where subtle morphological changes signal drug efficacy or toxicity.
Natural Language Processing (NLP)
Natural Language Processing (NLP) in biomedicine has come a long way from basic keyword searches to advanced models like BERT and BioGPT that understand context in scientific texts. These tools can do far more than just scan for terms; they can identify new mechanisms of action, summarize findings from clinical trials, and even flag potential off-target effects hidden in unstructured data. What is even more exciting is how NLP is now being combined with structured data to build biomedical knowledge graphs. These graphs link drugs, diseases, and targets in ways that can reveal unexpected therapeutic opportunities—connections that might be missed through traditional analysis.

Generative Models (GANs, VAEs, Reinforcement Learning)
Generative models are increasingly used for de novo molecular design, proposing novel chemical entities optimized across multiple objectives such as potency, selectivity, and ADMET properties. Variational autoencoders (VAEs) encode molecular structures into latent spaces, allowing interpolation and exploration of drug-like compounds, while GANs generate chemically valid structures with improved diversity. Reinforcement learning (RL) frameworks apply reward-based optimization strategies to tune generated molecules toward desired profiles, e.g., low toxicity or synthetic accessibility, based on predefined scoring functions.
Graph Neural Networks (GNNs)
GNNs treat atoms and bonds as nodes and edges in a graph, capturing molecular interactions more faithfully than string or image-based methods. They are now being extended to multi-modal systems that also incorporate protein graphs or gene interaction networks, allowing simultaneous modeling of drug and target structure. Recent advances include 3D-aware GNNs, which incorporate spatial geometry to better simulate ligand-protein binding.
Physics-Informed AI
Hybrid models that combine quantum mechanics and classical molecular dynamics with AI predictions improve the accuracy of force field estimations and binding free energy calculations. These models reduce computational costs typically associated with quantum chemical simulations, while preserving interpretability and physical plausibility. They are particularly useful in fragment-based drug discovery and predicting conformation-sensitive properties like chirality and polarization.
Multi-Task and Transfer Learning
Multi-task learning allows a single model to predict several endpoints simultaneously (e.g., activity against multiple targets, plus toxicity and permeability), enabling more efficient data use. Transfer learning adapts pretrained models, often trained on large, general chemical libraries, to new tasks with limited data, such as rare diseases or novel protein classes. This enables data-efficient innovation in therapeutic areas where experimental data is scarce or expensive to generate.
Active Learning
Active learning models help decide which compounds to test next by focusing on areas where the model is most uncertain. This smart, step-by-step approach leads to better experimental design and faster identification of promising candidates. Today, many platforms use active learning as part of automated, closed-loop systems, where real-time feedback from experiments helps the model improve continuously, without needing to generate massive amounts of data.
AIDD Across the Drug Discovery Pipeline
Target Identification and Validation
This is the foundational stage of drug discovery, where AIDD plays a transformative role by shifting from hypothesis-driven to data-driven approaches. AI integrates diverse omics datasets, such as genomics (gene mutations, expression profiles), proteomics (protein interactions, abundance), transcriptomics (RNA expression), and epigenomics, to build a multidimensional map of disease mechanisms. By employing unsupervised clustering, graph analytics, and machine learning models, AI reveals hidden regulatory networks and identifies novel, previously unrecognized therapeutic targets.
NLP algorithms extract structured knowledge from scientific literature, clinical trial registries, and public databases to strengthen hypotheses and validate molecular targets. More recently, AI has been applied to single-cell sequencing data to pinpoint target expression at the level of individual cell types—a critical capability in fields like oncology and immunology. By combining AI with functional genomics tools like CRISPR screens, researchers can now identify essential genes and synthetic lethality relationships. This helps prioritize drug targets not just for their novelty but also for their biological relevance and how realistically they can be pursued.
Hit and Lead Discovery
AI is speeding up hit discovery through high-throughput virtual screening, where millions of molecules are quickly evaluated for how well they might bind to a target. Machine learning models help guide molecular docking by predicting key properties like binding affinity, selectivity, lipophilicity, and ease of making a compound. Even when crystal structures are not available, AI can still enable structure-based screening using predicted protein models or homology-based approaches.

AI also supports ligand-based virtual screening by comparing pharmacophore features and 3D shapes to rank compounds. Generative models like VAEs and GANs can even design entirely new molecules tailored to a target's profile—balancing novelty, synthetic feasibility, and chemical diversity, with control over features like ring systems or chirality. Modern screening platforms now use multi-objective optimization to evaluate thousands of compounds not just for activity but also for off-target effects, toxicity risks, and drug-like properties, making early-stage discovery far more strategic than traditional high-throughput screening.
Lead Optimization
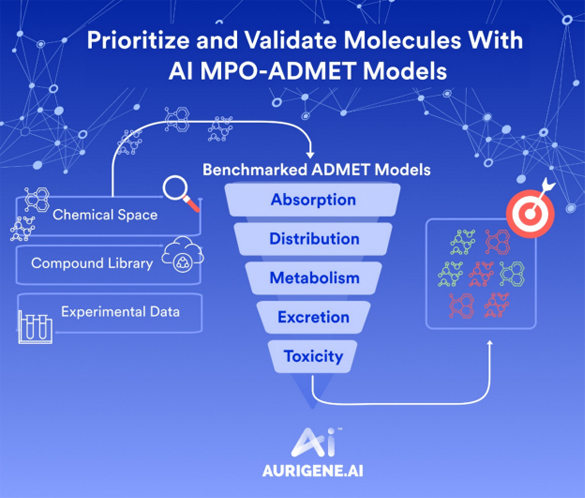
Once initial hits are identified, AI significantly enhances lead optimization by modeling structure-activity relationships (SARs) across multiple dimensions. Deep neural networks are trained to predict ADMET properties—absorption, distribution, metabolism, excretion, and toxicity—simultaneously with efficacy, guiding chemists to make data-informed structural modifications.
AI identifies subtle molecular features such as hydrogen-bonding capacity, polar surface area, and metabolic hotspots that influence bioavailability or clearance. RL and evolutionary algorithms are increasingly used in this stage to suggest iterative changes to lead structures that optimize potency while minimizing liabilities such as cardiotoxicity or poor permeability.
AIDD also supports synthetic route prediction, helping chemists evaluate how feasible it is to synthesize optimized compounds before actual lab work begins. This avoids wasting time on molecules that are overly complex or unstable. By incorporating physicochemical and pharmacokinetic profiles into real-time model updates, the optimization process becomes both faster and more likely to produce successful clinical candidates.
Preclinical Modeling
AI revolutionizes preclinical assessment by allowing in silico prediction of a drug's behavior in biological systems. ML models trained on large-scale in vivo and in vitro datasets can estimate a compound's pharmacokinetic profile, e.g., half-life, Cmax, clearance rates, and simulate tissue distribution using physiologically based pharmacokinetic (PBPK) models.
Toxicity prediction benefits from AI's ability to identify toxicophores and adverse outcome pathways directly from chemical structures. Deep learning tools can predict risks like hepatotoxicity, cardiotoxicity, genotoxicity, and immunotoxicity early in the pipeline, helping eliminate unsafe candidates upfront. AI also aids in selecting the most relevant animal models and study endpoints—or, in some cases, suggests skipping in vivo tests altogether when in silico evidence is strong. This approach reduces animal use, lowers costs, and improves translatability to human biology, especially when paired with data from organoids or organ-on-chip systems.
Clinical Trial Design
AIDD enhances clinical development through predictive patient stratification, which involves identifying subpopulations most likely to respond to a given therapy. AI models incorporate biomarker expression, genetic background, comorbidities, and lifestyle factors to match patients with optimal dosing regimens.
ML also helps simulate clinical trial outcomes based on prior data, guiding the selection of primary and secondary endpoints and adaptive trial designs. AI can flag likely drop-outs or adverse events early, improving retention and minimizing trial disruptions.

NLP of previous trial reports, real-world data (e.g., EHRs), and post-marketing surveillance enhances forecasting of patient response variability. Additionally, trial site selection is being optimized through AI analysis of geographic disease prevalence, investigator performance, and infrastructure readiness, contributing to faster recruitment and lower trial costs.
Drug Repurposing
AI has proven especially powerful in discovering new indications for existing drugs, reducing the time, cost, and risk associated with de novo drug discovery. Using data-driven modeling of gene expression signatures, disease phenotypes, and protein interaction networks, AI identifies overlapping biological pathways that may be influenced by drugs already on the market.
For example, knowledge graphs constructed by AI link molecular targets, clinical outcomes, and treatment guidelines to reveal hidden therapeutic opportunities. AI models have repurposed antivirals, kinase inhibitors, and anti-inflammatory agents across oncology, infectious diseases, and neurodegenerative disorders.
Advanced NLP is applied to mine case reports, trial registries, and observational studies for off-label successes and adverse effects, which are then incorporated into repurposing hypotheses. This has accelerated repurposing efforts during crises like COVID-19, where drugs like baricitinib and remdesivir were rapidly advanced into trials.
Data as the Critical Fuel
At the heart of AIDD is not just smart algorithms, it is the data that fuels them. These models are only as powerful as the information they are trained on. High-quality, diverse, and well-labeled datasets are what give AIDD systems their edge. Whether it is forecasting how molecules will interact or identifying the right patients for clinical trials, success depends on having strong, reliable data every step of the way.

Common data sources include:
Chemical Libraries
Repositories such as ChEMBL, ZINC, and PubChem provide millions of annotated small molecules with associated physicochemical properties, biological targets, and assay results. These databases are foundational for virtual screening, lead optimization, and QSAR modeling.
Binding and Structural Databases
Datasets like the Protein Data Bank (PDB) and BindingDB offer structural and affinity data for protein-ligand interactions, enabling AI to model binding conformations and predict activity with greater spatial accuracy.
Omics Repositories
Resources such as The Cancer Genome Atlas (TCGA) and GTEx provide multi-dimensional biological data—gene expression, methylation, mutation, and transcriptomic profiles—important for target identification and biomarker discovery. These datasets are particularly relevant for disease-specific modeling and stratification.
Clinical and Phenotypic Data
Platforms like ClinicalTrials.gov host vast records of trial outcomes, side-effect profiles, and response variability. This helps AI predict likely trial success or failure and enables smarter design of new trials.
Real-World Evidence (RWE)
Electronic Health Records (EHRs), adverse event databases (e.g., FAERS), insurance claims, and patient-reported outcomes offer invaluable post-marketing insights. These can be mined to detect long-term drug safety trends, support repurposing, and validate preclinical predictions in real populations.
The Challenge of Messy Reality
Despite the vast amount of biomedical and chemical data now available, AIDD faces several systemic challenges that hinder its full potential. One major issue is fragmentation and siloing, where valuable data is scattered across public databases, proprietary platforms, academic labs, pharmaceutical companies, and healthcare systems. This separation restricts the ability of AI models to generalize or learn across diverse sources. Additionally, data heterogeneity poses a significant challenge, as datasets differ widely in format, granularity, and terminology. Variations in assay protocols, molecular nomenclature, patient demographics, and data types—such as omics, imaging, and clinical records—create inconsistencies that can degrade model performance.
Another persistent obstacle is data sparsity and missingness, especially common in rare disease research and early-stage discovery. In such settings, datasets are often incomplete or insufficiently powered, making it difficult for standard AI methods to extract reliable insights without advanced imputation techniques or transfer learning. Moreover, noise and bias in data, introduced by annotation errors, batch effects in laboratory data, and selective reporting in the literature, can skew AI predictions, often in ways that are difficult to detect. Lastly, the lack of standardization across datasets further complicates integration. Without uniform schemas for molecular structures, assay results, or clinical outcomes, combining datasets becomes a labor-intensive process prone to errors and misinterpretations. These challenges collectively underscore the importance of data curation, harmonization, and collaborative frameworks in realizing the full promise of AI in drug discovery.
Innovations to Bridge the Gap
To overcome the data-related challenges that hinder AI-assisted drug discovery, several strategic, technical, and collaborative innovations have emerged. These aim to enhance data accessibility, quality, and integration while preserving privacy, standardization, and interpretability.
Federated Learning
Federated learning offers a transformative solution to both data access inequality and data siloing, which are major challenges in the pharmaceutical and healthcare industries. Instead of transferring sensitive data across institutional or national boundaries, federated learning keeps the data in its original location—such as a hospital, research institute, or pharmaceutical company—and sends only the trained model parameters or updates to a central aggregator. These updates are then combined to produce a global model that benefits from a broad range of input data without compromising security, patient privacy, or intellectual property.

This decentralized architecture is particularly important in regulated environments where frameworks like GDPR, HIPAA, and internal data-sharing agreements limit traditional data pooling. In the context of AIDD, federated learning supports collaborative model training across drug companies, hospitals, and research labs while preserving data sovereignty and trust. By enabling access to diverse chemical, clinical, and biological datasets across silos, federated learning produces more robust and generalizable models that reflect real-world biological and demographic variation—ultimately enhancing discovery while maintaining compliance and confidentiality.
Data Fusion and Harmonization Pipelines
To address data heterogeneity and fragmentation, data fusion frameworks are used to integrate information from multiple sources, such as chemical libraries, gene expression datasets, and clinical trial records, into a unified, interoperable format. These pipelines use schema mapping, data normalization, and ontology alignment techniques to ensure consistency. Ontologies like MeSH, UMLS, and SNOMED CT help map equivalent terms across domains (e.g., connecting "myocardial infarction" with "heart attack"). In probabilistic merging, algorithms estimate the likelihood that similar entries refer to the same biological entity or patient, thereby reducing duplication and inconsistency. Consequently, AI models can operate on multi-modal, cross-source inputs, enabling more robust predictions and broader insights than siloed data would permit.
Synthetic Data Generation
In data-scarce domains like rare diseases, early-stage compounds, or pediatric populations, AI-driven synthetic data generation offers a vital solution. Generative models such as GANs and VAEs simulate realistic patient data, chemical structures, or assay results that mirror real-world patterns without duplicating actual data.
Automated Curation Tools
Manual data curation is a bottleneck in biomedical research owing to the volume and complexity of the data. To address this, automated curation tools powered by NLP and machine reading comprehension are now being deployed. These tools can scan vast scientific corpora, including journal articles, patents, conference proceedings, and clinical case reports, to extract relevant facts, annotate drug-target relationships, and standardize terminology. Some systems also validate extracted data by cross-referencing against structured databases. This automation not only improves speed and scalability but also enhances accuracy and completeness, especially in areas like pharmacovigilance, where timeliness and context are critical for assessing drug safety signals.
Global Consortia and Benchmarking Efforts
Cross-sector initiatives are driving collaboration and standardization in AIDD. One key example is MELLODDY (Machine Learning Ledger Orchestration for Drug Discovery), a multi-pharma consortium that enables collaborative model development using federated learning across more than a billion molecules—all without exposing proprietary data. Additionally, the adoption of the FAIR principles—that data should be Findable, Accessible, Interoperable, and Reusable—has become a cornerstone of open science and shared infrastructure efforts. These global frameworks promote consistent data formatting, transparent methodologies, and ethical AI usage, accelerating discovery and reducing duplication of effort across the pharmaceutical ecosystem.
Model Validation with Ground Truth Benchmarks
AI models are only as good as their ability to perform consistently across independent, unbiased test sets. To that end, benchmarking datasets such as MoleculeNet, Tox21, and SIDER have become standard in evaluating and comparing AIDD tools. These curated datasets come with clearly defined tasks, such as toxicity prediction, solubility classification, or bioactivity scoring, along with metrics like ROC-AUC, precision-recall, and F1 score. Benchmarking not only improves reproducibility and transparency but also drives innovation by highlighting gaps where current models underperform. Some benchmarks now incorporate time-stamped test sets or domain-shift scenarios to simulate real-world conditions more accurately.
Together, these innovations represent a convergence of AI, informatics, privacy engineering, and biomedical collaboration, all designed to transform fragmented, inconsistent, and siloed datasets into fuel for trustworthy, scalable, and impactful AI-driven drug discovery.
What Aurigene offers
Aurigene has developed a state-of-the-art platform, Aurigene.AI, to transform the pace, accuracy, and success rate of modern drug discovery. With over two decades of experience across therapeutic discovery and development, Aurigene is uniquely positioned to integrate cutting-edge AI technologies with experimental capabilities, offering a true end-to-end AIDD solution. The company's innovation is centered on addressing the limitations of traditional drug development processes by embedding AI tools into every critical phase of discovery and preclinical decision-making.
At the heart of Aurigene.AI lies a meticulously curated and dynamically expanding compound database. This powerful resource comprises over 180 million compounds and 1.6 million validated bioassay data points, carefully collected from both in-house experiments and public domain sources. The database undergoes structure normalization and quality validation, ensuring consistency and reliability across all data points. It functions as both a search engine for researchers and a training bed for AI models, supporting structure-based and property-based queries as well as semantic search. By offering access to deeply annotated, high-quality chemical and biological data, the database lays the groundwork for robust, data-driven decision-making at all stages of drug discovery.
Aurigene.AI provides an integrated and modular environment that combines AI with computational chemistry to facilitate faster and smarter decision-making. The platform currently houses 11 generative AI models, 4 predictive AI models, and 8 computational/CADD modules, each focused on different aspects of drug design and optimization. Generative models are used for novel scaffold generation and molecular ideation; predictive models estimate properties such as ADMET and efficacy; and CADD modules support structure-based modeling, docking, and simulations. The modular architecture allows scientists to customize model selection according to specific project needs, making it a comprehensive and flexible digital ecosystem capable of supporting everything from hit generation to candidate nomination.
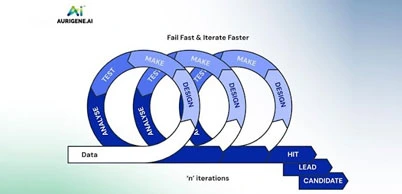
A distinguishing feature of Aurigene's platform is its ability to generate, test, and refine molecules in a continuous cycle. Once AI models shortlist or design molecules based on multiple parameters, such as potency, selectivity, and pharmacokinetics, those molecules are rapidly synthesized and subjected to experimental validation. The results from in vitro and in vivo testing are fed back into the system in real time, retraining the AI models to improve their future predictions. This closed-loop feedback mechanism ensures that every iteration leads to better-optimized molecules with higher success potential. It also accelerates the identification of failures, allowing researchers to pivot early and efficiently reinitiate the design process. The outcome is a faster, smarter, and more reliable route to high-quality drug candidates.

Unlike standalone AI tools, Aurigene.AI is deeply embedded in Aurigene's broader experimental infrastructure. Once the AI generates viable molecules, Aurigene's synthetic chemistry team can quickly produce them for laboratory evaluation. The platform is directly connected to Aurigene's discovery biology capabilities, which include a broad range of in vitro and in vivo assays. These assays validate the AI's predictions, assess efficacy and toxicity, and support mechanism-of-action studies. Importantly, the integration of digital and experimental workflows allows for rapid turnaround, continuous learning, and adaptive experimentation. This combination of in silico prediction and real-world validation makes Aurigene's AIDD platform truly translational and practical.
To complement its design capabilities, Aurigene has also built Aurimine, a proprietary virtual compound library with over 12 billion structures. This vast chemical space is searchable and usable in conjunction with generative AI tools for tasks such as ultra-large virtual screening, scaffold hopping, diversity analysis, and lead expansion. Aurimine empowers discovery teams to explore unprecedented molecular diversity while maintaining high chemical relevance and synthetic accessibility. The ability to screen billions of virtual molecules dramatically increases the chances of identifying novel hits that are both unique and developable.
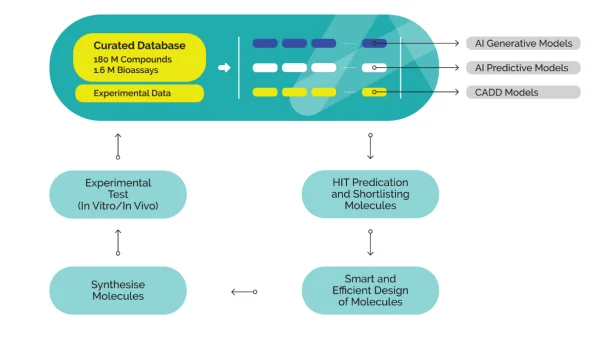
Aurigene's AIDD ecosystem is strategically designed to address several longstanding bottlenecks in drug discovery, namely, high failure rates, slow development timelines, and unsustainable R&D costs. By fusing AI with experimental execution, Aurigene offers a streamlined, intelligent discovery process that supports rapid candidate triaging, informed molecular design, and early-stage risk mitigation. The platform's adaptability and modularity allow researchers to pursue diverse therapeutic targets while maintaining scientific rigor and operational speed. Moreover, the integration of feedback from synthesis and bioassays makes every AI-driven design cycle more intelligent than the last. This ability to evolve rapidly with each iteration significantly increases the probability of success while minimizing development costs and timelines.
Emerging Trends and Innovations
AI is poised to revolutionize drug discovery, offering transformative potential across various facets of the pharmaceutical landscape.
Medicinal Chemistry Integration
AI is rapidly becoming an integral part of medicinal chemistry, transforming the way chemists design, analyze, and optimize drug candidates. Advanced ML models assist chemists in real time by proposing structural analogues, forecasting retrosynthetic routes, and suggesting bioisosteric replacements that maintain pharmacological activity while improving drug-like properties. These AI tools accelerate iterative design by reducing reliance on manual SAR analysis and increasing the speed of hypothesis testing. Moreover, molecular generative models now incorporate synthetic feasibility filters, ensuring that AI-proposed molecules are not only potent but also synthetically tractable. In practice, this integration has led to shorter design cycles and more innovative lead series, making AI a true "co-designer" in the medicinal chemistry workflow.
Closed-Loop Systems
One of the most promising advancements in AIDD is the development of closed-loop discovery systems that integrate AI with robotic automation and real-time data capture. In this model, molecules are designed by AI, synthesized via automated chemistry platforms, and tested in high-throughput assays. The resulting biological data is then immediately fed back into the AI system, retraining the models to refine subsequent iterations. This loop allows for self-improving, adaptive optimization, drastically reducing the delay between design and experimental validation. Emerging platforms even use Bayesian optimization and reinforcement learning to guide exploration, focusing on the most informative molecules in chemical space. Such systems have demonstrated the potential to compress early-stage discovery timelines from years to months, creating a paradigm shift toward continuous, data-driven drug design.
Multi-Modal and Multi-Omics AI
Traditional drug discovery often treats data sources (e.g., molecular structures, patient biomarkers, imaging scans) as siloed. However, multi-modal AI models are designed to process and integrate heterogeneous data types, combining chemical, biological, clinical, genomic, transcriptomic, and imaging data into unified predictive frameworks. For instance, a single model might correlate a compound's chemical structure with its cellular phenotype and the genetic background of a patient population. This holistic view enhances the ability to predict not only efficacy and safety but also patient-specific response profiles, paving the way for precision medicine. Multi-modal learning is particularly impactful in oncology, where genomic mutations, immune contexture, and tumor histology jointly influence treatment outcomes. By capturing complex interdependencies, these models improve translational accuracy and clinical relevance.
Rare and Neglected Diseases
Drug discovery in rare and neglected diseases has long suffered from a lack of financial incentives and sparse data. AI, particularly transfer learning and few-shot learning, is now helping bridge this gap. Models trained on rich datasets from well-studied conditions can be fine-tuned to predict outcomes for low-data diseases, reusing latent knowledge without needing thousands of examples. Additionally, synthetic data generation and generative modeling techniques are being used to simulate plausible compound-target interactions where experimental data is scarce. These innovations lower the barrier to entry for rare disease R&D and are beginning to attract interest from non-profits, academic centers, and even commercial drug developers. By reducing development risk and cost, AIDD is making discovery for small patient populations both scientifically and economically feasible.
Pandemic Preparedness
The COVID-19 pandemic spotlighted the urgent need for rapid-response therapeutic discovery platforms. AI pipelines proved invaluable during the crisis by enabling drug repurposing, virtual screening, and target identification within weeks—efforts that traditionally would have taken months or years. Going forward, AI platforms are being formalized as core infrastructure for future pandemic preparedness. This includes the ability to mine emerging pathogen genomes, predict host-pathogen interactions, simulate immune responses, and identify cross-reactive compounds or vaccine targets. Several governments and global health organizations are now investing in pre-trained AI frameworks and cloud-based collaboration hubs that can be activated in emergency settings. In essence, AIDD has transitioned from a research luxury to a public health necessity, offering scalable, rapid, and adaptive drug discovery tools for global crises.

Ethics and Policy
As AI becomes embedded in high-stakes biomedical decisions, ethical and policy frameworks are urgently evolving to ensure responsible use. Concerns around algorithmic bias, data privacy, and decision transparency have prompted calls for explainable AI (XAI), especially in clinical-facing applications. Models must be interpretable to regulators, physicians, and patients alike—particularly when used for trial recruitment or dosing optimization. Additionally, there is growing awareness of data inequity, where models trained predominantly on data from high-income populations may underperform in underrepresented groups. Global regulatory bodies, including the FDA and EMA, are actively developing guidelines to assess AI/ML systems used in drug development. Meanwhile, the FAIR (Findable, Accessible, Interoperable, Reusable) and TRUST data principles are being incorporated into AI model documentation, ensuring reproducibility and auditability. Policy discussions also include intellectual property rights for AI-generated molecules and accountability mechanisms for AI errors or adverse events.
Challenges and Bottlenecks
Interpretability
One of the most persistent challenges in AIDD is the opacity of complex AI models, especially deep learning systems that utilize thousands or even millions of parameters. These models often make highly accurate predictions but fail to provide clear, understandable explanations for their outputs, which is a major barrier in a field where scientific reasoning, traceability, and regulatory scrutiny are essential.
In drug discovery, it is not enough for a model to suggest a molecule or a target; stakeholders must understand why a particular prediction was made, especially when human lives are involved. Current post-hoc methods like SHAP, LIME, and attention visualization offer partial solutions, but they are not yet robust or standardized across platforms.
The inability to fully trace decision pathways hinders adoption in regulated environments and limits scientific confidence in AI-generated hypotheses, especially when novel targets or chemotypes are proposed without existing literature support.
Validation Gap
AI models are only as useful as their ability to produce experimentally verifiable results. However, there remains a significant gap between computational predictions and biological confirmation. Even when AI accurately forecasts binding affinity or ADMET properties, the downstream validation through synthesis, assay development, and in vivo studies is labor-intensive, costly, and sometimes technically unfeasible. This is particularly problematic in early-stage discovery where lab resources are limited, and throughput is constrained.
Additionally, novel AI-designed compounds often push into unexplored chemical space, where existing assays or animal models may not yet be optimized—introducing further uncertainty. The disconnect between in silico potential and practical feasibility can lead to delays in progressing otherwise promising candidates and creates skepticism among bench scientists who rely on empirical reproducibility.
Regulatory Adaptation
While regulatory agencies are increasingly open to AI's role in drug development, the absence of clear, harmonized frameworks for evaluating AI-generated evidence remains a roadblock. Existing drug approval pathways are built around human hypothesis generation, well-defined endpoints, and controlled experimentation, not black-box optimization. Questions around data provenance, model versioning, auditability, and long-term performance monitoring remain largely unresolved.
There is also ambiguity regarding the evidentiary weight AI predictions should hold when submitted alongside traditional clinical data. Regulatory uncertainty can deter pharmaceutical companies from fully embracing AIDD, especially in critical decisions like IND filings or biomarker qualification. Furthermore, the global regulatory landscape is fragmented: what is permissible under one authority may not be accepted by another, complicating multinational development programs and delaying market access.

Talent and Infrastructure
Deploying and scaling AIDD requires a convergence of skill sets rarely found within a single team. Effective implementation demands experts in AI/ML, cheminformatics, systems biology, software engineering, and drug development—all collaborating under a unified workflow. Many pharmaceutical organizations and academic labs struggle to attract and retain talent with cross-disciplinary fluency. Moreover, the computational demands of training and deploying high-resolution AI models, especially those incorporating generative chemistry, 3D molecular dynamics, or multi-modal inputs, require access to high-performance computing (HPC) environments, GPUs, cloud infrastructure, and secure data pipelines. Such infrastructure is not evenly distributed, especially among startups, smaller biotechs, and institutions in low- and middle-income countries. This asymmetry risks deepening the innovation divide, where only a few well-resourced organizations can fully exploit the potential of AIDD.
Future Outlook
Explainable AI (XAI)
The next generation of AI tools in drug discovery will be built with explainability as a core feature, not an afterthought. As regulatory bodies and clinicians increasingly scrutinize algorithmic recommendations, models must evolve to offer interpretable outputs, such as visual cues on molecular features responsible for predicted activity or clear explanations for target prioritization.
Researchers are now designing AI architectures that include built-in attention mechanisms, counterfactual reasoning, and saliency mapping, enabling end-users to trace how predictions are formed. In drug discovery contexts, XAI will support critical decision points—such as lead selection or toxicity flagging—by offering transparent justifications. This enhances not just regulatory readiness but also fosters human-machine trust, especially in cross-functional teams where clinicians, chemists, and data scientists must align their interpretations of model behavior.
Global Standardization
One of the key barriers to industrial-scale adoption of AIDD is the lack of universally accepted standards for data formatting, model validation, and performance benchmarking. To overcome this, the field is moving toward the establishment of open-source repositories, annotated benchmarks, and transparent evaluation protocols that allow researchers and regulators to assess AI systems on a level playing field. Initiatives like MoleculeNet, Tox21, and Open Targets are laying the groundwork, but further standardization is needed in areas like ADMET prediction, multi-target modeling, and clinical endpoint simulation. International consortia, including IMI and ICH, are also beginning to incorporate AI-readiness frameworks into drug development guidelines. Standardization not only ensures model comparability and reproducibility, but it also accelerates regulatory convergence—critical for global submissions.
Democratization
The future of AIDD depends not only on technological advancement but also on accessibility and equity. Democratization efforts aim to level the playing field by expanding access to powerful AI tools, curated datasets, and scalable infrastructure through open-source software, cloud-based platforms, and low-code environments. Startups, academic labs, and researchers in low-resource settings can now leverage platforms like DeepChem, OpenChem, and cloud-hosted Jupyter notebooks to build and test AI models without needing enterprise-level computing power.
Democratization also means upskilling the workforce, through training modules, workshops, and integration into graduate curricula, to ensure that scientists and clinicians alike can confidently use AI tools. These efforts are essential for fostering global innovation, inclusivity, and diversity of thought in drug discovery.
Human-AI Synergy
Rather than replacing human scientists, the long-term vision of AIDD is one of collaborative intelligence, where AI augments human decision-making by offering rapid insight generation, cross-dataset pattern recognition, and simulation at scale. The most effective discovery teams will not be fully automated but rather composed of hybrid systems in which AI handles hypothesis generation, risk assessment, and optimization, while humans guide interpretation, intuition, and ethical judgment. This synergy is already emerging in workflows where AI flags lead candidates, and chemists validate and redirect based on domain knowledge. Over time, we can expect AI interfaces to become more interactive, allowing for real-time dialogue between scientists and models, creating a continuous learning loop where both parties—human and machine—evolve together.

The future of AI-Assisted Drug Discovery is not a distant vision—it is unfolding now, marked by a confluence of ethical awareness, technological maturity, and interdisciplinary collaboration. As explainability improves, federated learning expands, and democratized platforms proliferate, AIDD will become a standard part of pharmaceutical innovation pipelines. Its potential to reduce development time, lower costs, improve target selection, and personalize therapy is no longer theoretical. By addressing the remaining barriers and embracing collective innovation, AIDD stands poised to redefine how we discover, develop, and deliver medicine—faster, fairer, and more intelligently than ever before.